A chewable multivitamin has been recalled across 25 states following an inspection by the U.S. Food and Drug Administration (FDA), which found that the product contained lower-than-intended levels of vitamin D. The recall affects the Multivitamin with Fluoride Chewable Tablets in grape flavor, manufactured by Winder Laboratories, LLC, located in Winder, Georgia.
This recall involves two specific versions of the multivitamin. One version contains 0.25 mg of fluoride, while the other contains 1.0 mg. Both products are sold in bottles of 100 tablets. The identification details are as follows: the NDC 75826-169-10 and UPC 3 15826 16910 2 for the 0.25 mg variant, and the NDC 75826-171-10 and UPC 3 15826 17110 5 for the 1.0 mg variant.
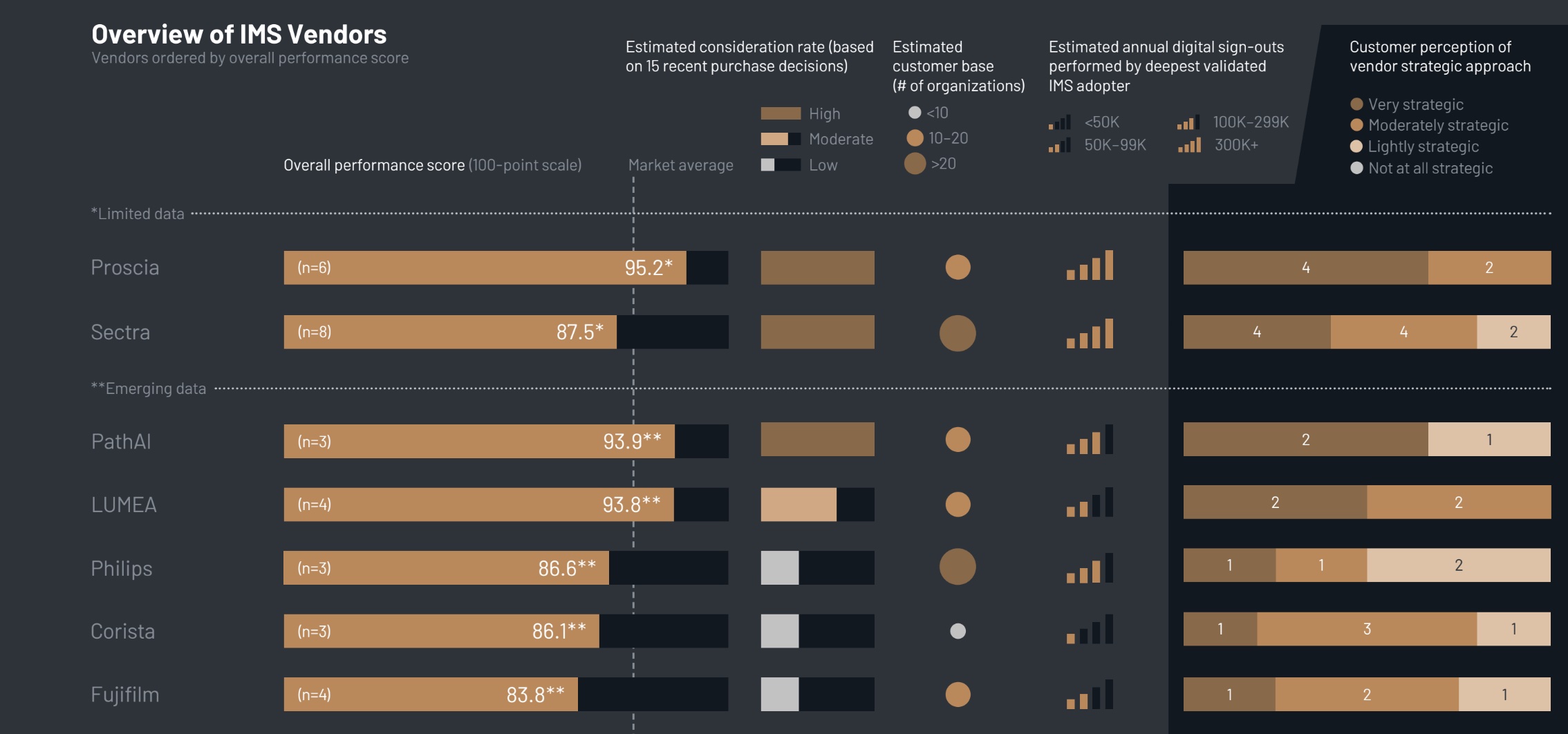
Details of the Recall
The FDA’s inspection revealed that the multivitamin was subpotent for vitamin D, meaning it contained less of this essential vitamin than specified. Vitamin D is crucial for bone health as it aids in calcium absorption and supports the immune system. Maintaining adequate levels of vitamin D is important for overall health, particularly for bones and muscles.
The voluntary recall was initiated by Winder Laboratories on October 27, 2025, after the inspection results were confirmed. According to FDA records, the recall affects 422 cases of the 0.25 mg product and 210 cases of the 1.0 mg product, with each case containing 12 bottles of 100 tablets.
Affected products can be identified by the following lot information:
– Lot 1692303, serial number 138909557498, expiration date October 26, 2025
– Lot 1692304, serial number 131163901709, expiration date October 26, 2025
– Lot 1712301, serial number 163590222021, expiration date November 14, 2025
The recall is documented under recall number H-0569-2026 and is associated with FDA Event ID 97884. Initial notifications regarding the recall were sent by letter to distributors.
Distribution Impact
The affected multivitamin was distributed to a wide range of states and territories, including Alabama, Arizona, California, Florida, Hawaii, Iowa, Illinois, Indiana, Massachusetts, Michigan, Missouri, Mississippi, North Carolina, New Jersey, New York, Oregon, Pennsylvania, Puerto Rico, Rhode Island, Tennessee, Texas, Utah, Washington, Wisconsin, and West Virginia.
The FDA has classified this recall as Class III, the lowest risk category. This classification indicates that exposure to the product is not likely to cause adverse health consequences, but it still violates FDA regulations. The FDA’s assessment suggests that the subpotency issue does not pose a significant health risk, while corrective actions remain necessary.
As of now, the recall is ongoing, and no termination date has been provided by the FDA. Consumers who may have purchased the affected products are advised to check their supplies and follow the recall instructions provided by the manufacturer.